INFORMATION PROVIDED BY
Chemical
Compounds
DEEP
PROFILING
SERVICES
For Any Chemical Compound comprising C, H, N, O, S, F, Cl, Br, I, Si, P, and/or As.
Full List of CC-DPS Information- Outline
- Chemical Identifiers
- Downloadable Files
- Constant Properties
- Temperature Dependent Properties
- Spectroscopic Analyses
- 3D Visualization, Animation and Analysis
- Quantum Chemical Information
- Molecular Descriptors
Outline
This page offers guidance on accessing the CC-DPS (Chemical Compounds Deep Profiling Services) information via our web-based user interface. Information is made available in various formats, including downloadable data files, data tables, charts, and images, ensuring comprehensive support for your research or application needs. For detailed information on locating and utilizing these resources, please refer to the sections below.
Chemical Identifiers
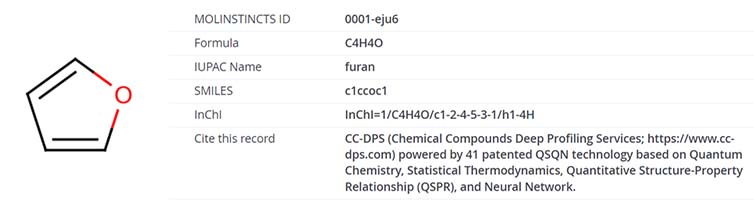
Each chemical compound in the CC-DPS information is presented with a distinctive 2D structure image and a unique Mol-Instincts ID (‘Mol-Instincts’ refers to the CC-DPS database system). Identifiers such as the compound’s formula, SMILES string, and InChI are furnished to ensure accurate recognition of the target compound. A citation format is available for referencing the CC-DPS information in academic or professional contexts. Displayed below is a sample record for the compound furan.
Downloadable Files
Below the chemical identifiers, you will find four types of downloadable files produced based on our QSQN technology, each serving a distinct purpose:

1. Physical Property Datafile (IKC) for Process Simulators (e.g. Aspen Plus): This IK-Cape (IKC) file contains thermophysical property data formatted for integration with process simulators like Aspen Plus. It is particularly useful when the simulator's built-in database lacks the component data you require. More information and a user guide for the IKC file are available on a dedicated webpage.
2. Full Molecular Descriptor File (CSV): For comprehensive analysis, we offer a complete set of over 2,000 molecular descriptor values for the targeted compound. This data is available in CSV file format for ease of use in various computational platforms. Additional details can are available on a dedicated webpage.
3. Quantum Chemical Computation Result File (FCHK): The formatted checkpoint (FCHK) file is a result of quantum chemical computations. It is an essential tool for researchers looking to enhance the precision and efficiency of their quantum chemical calculations. Additional details can be found on its dedicated webpage.
4. Quantum Chemically Optimized 3D Structure File (MOL): This file provides the optimized chemical structure data in MOL format, ascertained through quantum chemical computations for the respective compound, enabling precise modeling and analysis in various applications.
Constant Properties
Beneath the downloadable files section, you will find seven tabs categorized to cover each segment of the CC-DPS offerings. Within the ‘Constant Properties’ tab, 34 constant properties related to thermo-physicochemical, thermodynamic, and transport aspects determined based on our QSQN technology are provided.
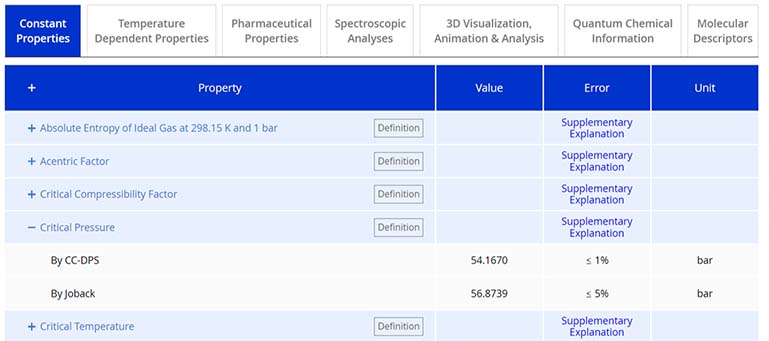
Expanding a property detail by clicking the “+” icon provides value in the 'Value' column determined by CC-DPS and by other existing approaches if applicable. Properties also evaluated by other existing approaches are listed in the “Other Existing Approaches” section on the Reliability page. The 'Error' column displays the deviation percentage from the refined experimental data, if available. Additionally, a 'Supplementary Explanation' link for each property offers an in-depth context for the error values listed.
Similarly, 16 constant pharmaceutical properties are provided and can be accessed under the “Pharmaceutical Properties” tab.
Temperature Dependent Properties
Within the 'Temperature Dependent Properties' tab, users can access 11 properties that vary with temperature, all of which are produced based on our QSQN technology. They are presented in both graphical and tabular formats. For instance, the heat capacity of liquid, showcased below, demonstrates how data are visualized:
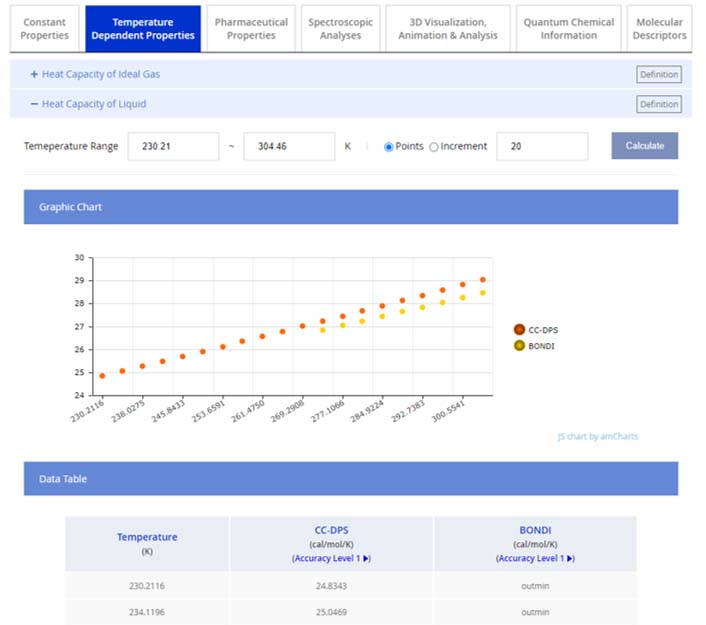
20 data points spanning a predetermined temperature range are displayed. Users have the option to specify a custom temperature range, which must align with the phase of the property, i.e., the chosen range for liquid properties should fall within the temperature at which the compound remains liquid.
Data provided by CC-DPS, along with data from other existing methods where applicable, are available for comparison. Properties also evaluated by other existing approaches are listed in the “Other Existing Approaches” section on the Reliability page. Each set of data includes an indication of the Accuracy Level, which is based on the refined experimental data when available. A link to the 'Accuracy Level' offers detailed context for the accuracy designation provided.
Spectroscopic Analyses
The 'Spectroscopic Analyses' tab features detailed charts and data tables for Infrared (IR) spectra, Nuclear Magnetic Resonances (NMR for C, H, N, O, S), and Vibrational Circular Dichroism (VCD) spectra. These are obtained from the quantum chemical computations described in our QSQN technology. By expanding the spectra details via the “+” icon, users can view the spectral chart and access the associated data set, exemplified by the IR spectra below.
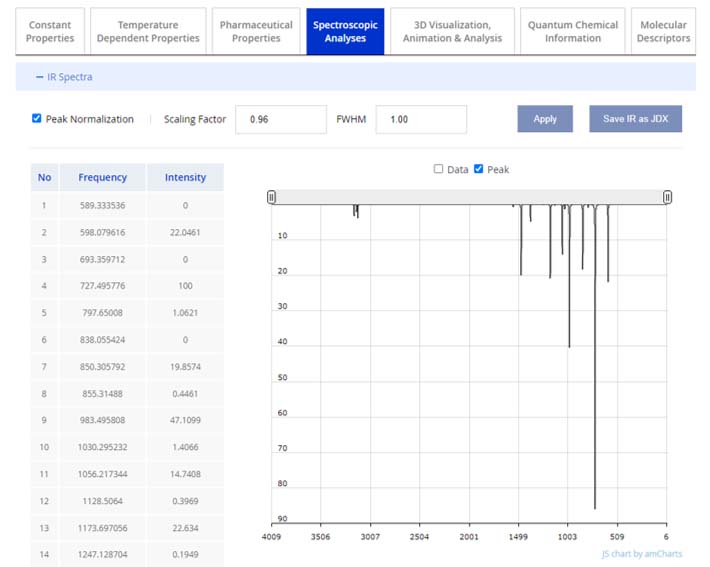
The interactive chart enables users to scrutinize the spectra through zoom-in and zoom-out features for a detailed or an overview perspective. Functionality includes peak normalization, scaling factor adjustments, and the Full Width at Half Maximum (FWHM) setting. The scaling factor is preset to 0.96, a value calibrated through comparison with an extensive dataset of over 2,500 experimental frequencies. For user convenience, the IR spectroscopy data is available for download in the JDX (JCAMP Chemical Spectroscopic Data Exchange Format) file format.
Vibrational animations related to IR spectroscopy are accessible in the ‘3D Visualization, Animation, & Analysis’ tab, detailed further in the following section.
3D Visualization, Animation and Analysis View Video Guide
Information, save, and copy: Information, Save, and Copy: Within the '3D Visualization, Animation, & Analysis' tab, you will initially see the structure of the target compound, optimized by quantum chemical computations, in the ‘Information, save, and copy’ menu section on the left side.
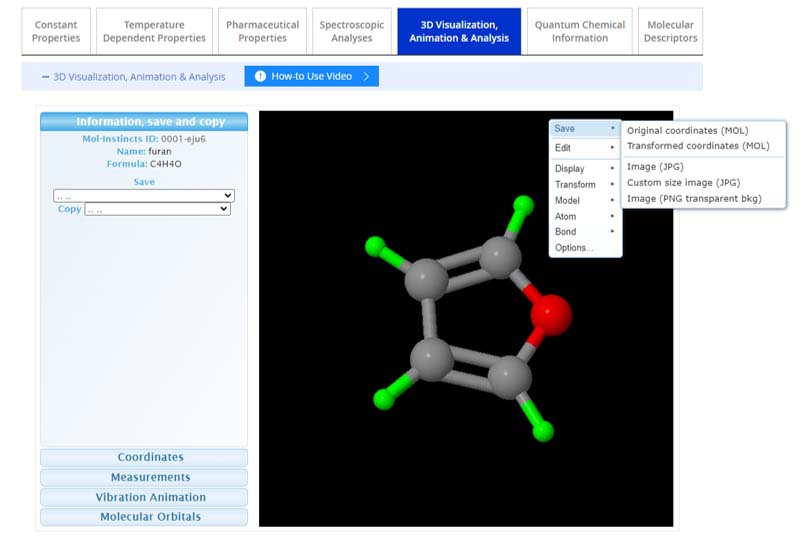
The optimized 3D structure on the right can be rotated interactively by keep clicking and moving the mouse button. Mouse wheel zoom is available as well – the size of the molecule can be increased or decreased by scrolling the mouse wheel. The image and the structure data of the molecule can be saved or copied to clipboard by using the menus in the left or menus appeared by right-click of the mouse.
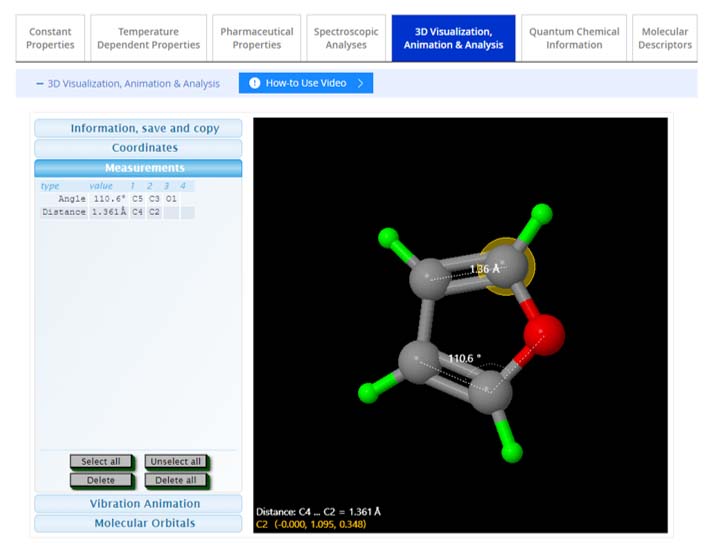
Coordinates, Measurements: The 'Coordinates' function provides the X, Y, Z coordinates of atoms within the 3D structure, which can be highlighted by selecting them on the model. The 'Measurements' feature allows users to determine bond lengths, angles, and dihedral angles. To measure distances, angles, and dihedral angles within the molecular structure: Double-click the start and end atoms and single-click any atoms in between.
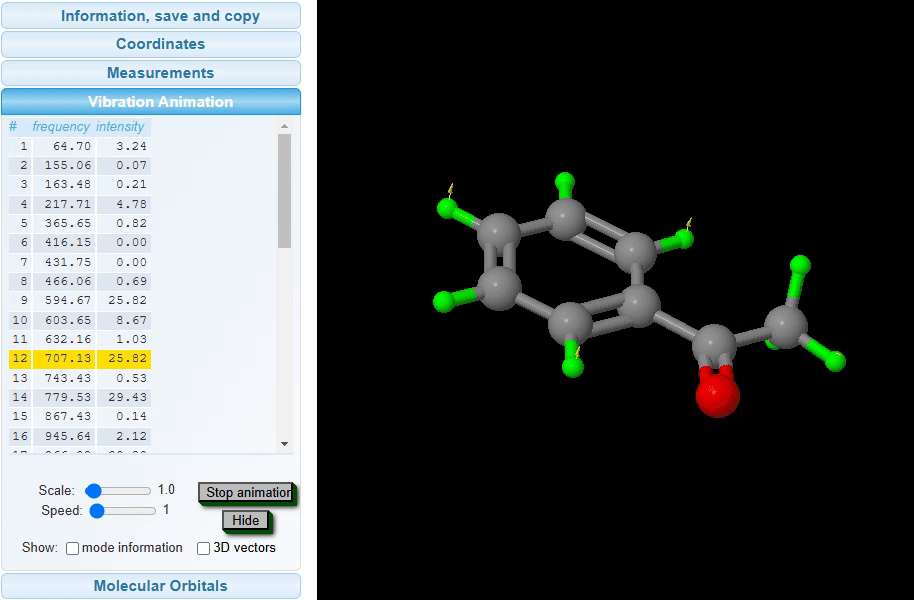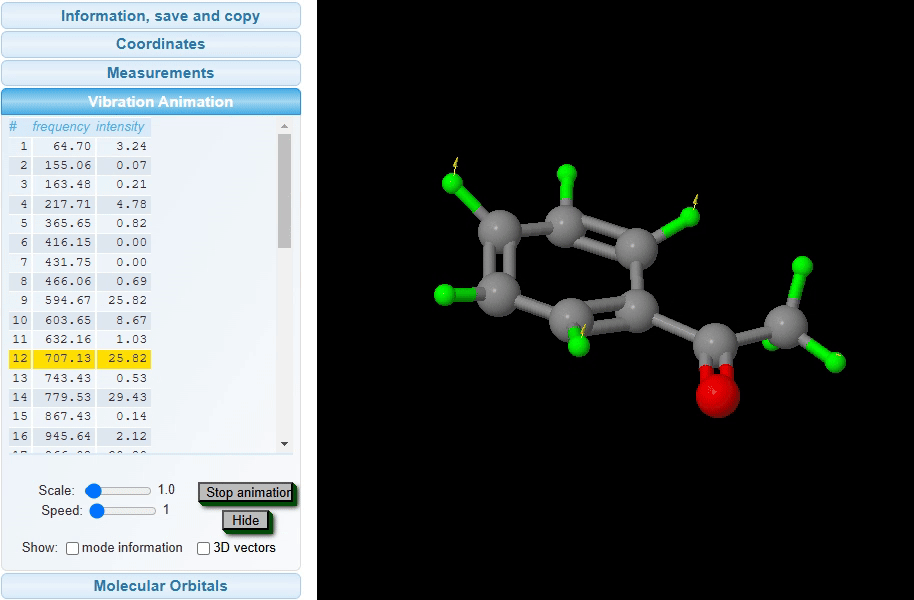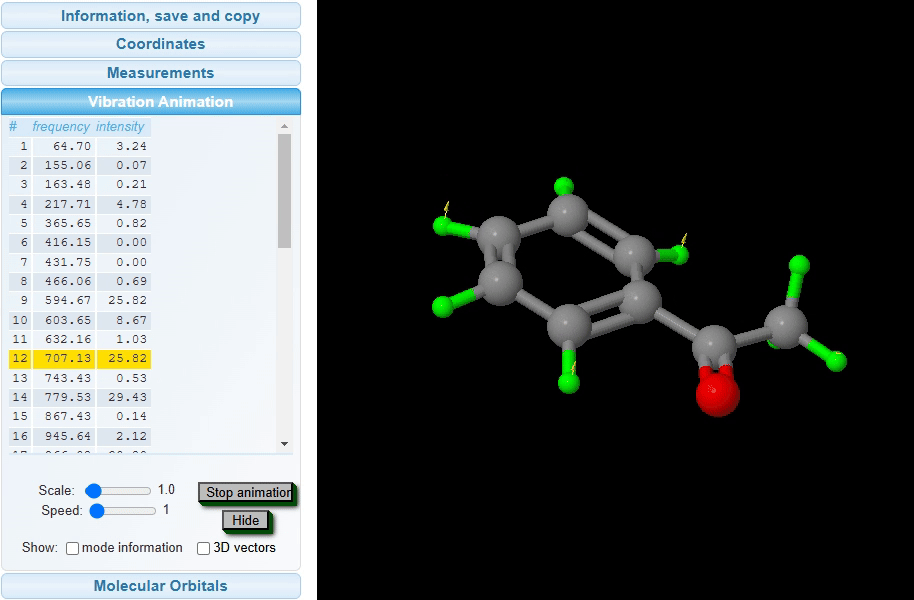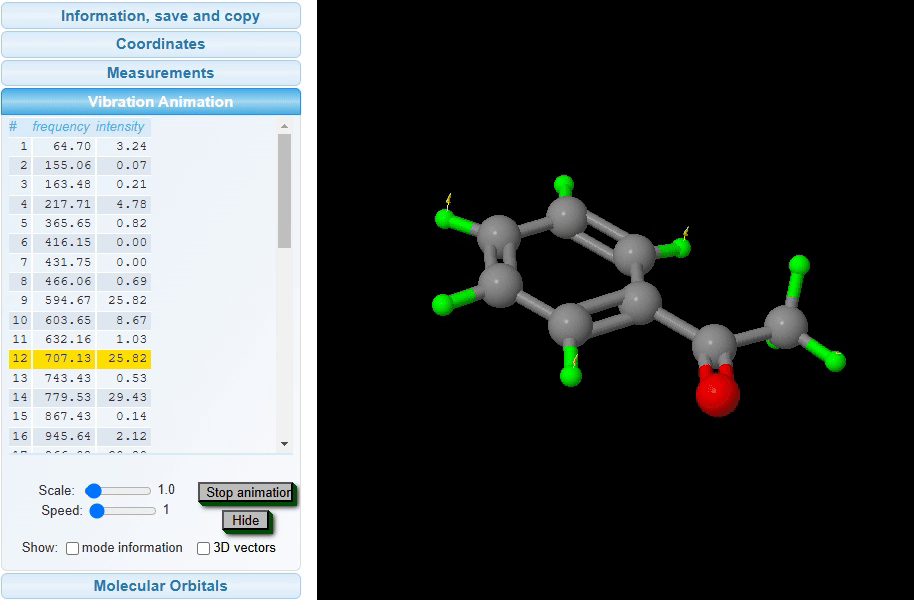
Vibration Animation: The vibrational animation feature visualizes the dynamic movements of atoms in a molecule at specific vibrational frequencies and intensities, as shown in the image below. By selecting a frequency from the list, users can view an animated representation of the molecular vibrations associated with that frequency. The intensity of these vibrations is reflected in the animation’s amplitude, providing an intuitive understanding of the vibrational energy levels present within the molecule.
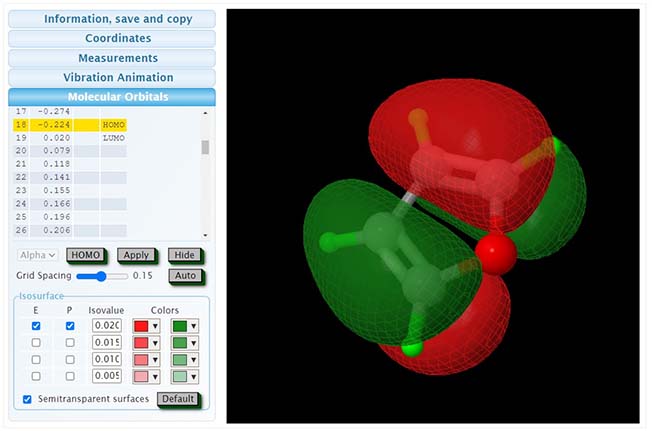
Molecular Orbitals: The Molecular Orbitals feature offers an interactive visualization of the molecule's electron distribution, including the Highest Occupied Molecular Orbital (HOMO) and Lowest Unoccupied Molecular Orbital (LUMO), as well as a full range of orbitals from HOMO-1, HOMO-2 to LUMO+1, LUMO+2, etc. The energy levels associated with each orbital are provided as well. The image illustrates below is the HOMO image of the target molecule. This tool enables users to not only visualize the 3D electron density clouds for each molecular orbital but also to understand the energetic hierarchy and the possible transitions within the molecular electronic structure.
Quantum Chemical Information
Under the 'Quantum Chemical Information' tab, users are provided with a curated selection of key data extracted from our quantum chemical computations. This includes essential figures such as total energy, counts of alpha and beta electrons, cartesian coordinates alongside gradients and force constants, dipole moments, and Mulliken charges.

For a more in-depth analysis, the full spectrum of quantum chemical data can be accessed and downloaded in the FCHK (formatted checkpoint) file format from the 'DOWNLOADABLE FILES' section, offering a complete set of computational results for advanced research purposes.
Molecular Descriptors
Within the 'Molecular Descriptors' tab, users can access an extensive array of over 2,000 molecular descriptors, organized into 24 distinct categories for comprehensive analysis. This assembly features a broad spectrum of critical descriptors from constitutional and geometrical to advanced 3D-MoRSE and WHIM, along with GETAWAY descriptors such as atom-centered fragments, and various indices including topological, connectivity, information, and autocorrelation indices.

The collection also includes advanced quantum chemical and electrostatic descriptors that are vital for QSAR/QSPR modeling, offering unparalleled insights into molecular characteristics. Derived from in-depth quantum chemical computations, these specialized descriptors are instrumental in the accurate prediction of molecular behavior and are crucial for robust predictive modeling in cheminformatics and materials science.
For those engaged in granular analysis, the complete set of molecular descriptor values is available for download as a CSV file in the 'Downloadable Files' section, providing a rich dataset for advanced scientific research and development applications.